Research
Models of evolutionary processes help us understand how biodiversity is generated, sustained, and lost.
My primary research interests include statistical phylogenetics, Bayesian inference, historical biogeography, and phenotypic evolution.
The Landis Lab studies evolution by developing probabilistic models, writing open source and community-minded software, and analysing simulated and empirical data.
These methods are available as open source software (see below).
We maintain a broad and active interest in phylogenetic inference, and develop for RevBayes, an open-source package for modeling evolutionary processes and estimating trees.
Historical biogeography
Historical biogeography studies the distribution of species in space and time.
Because many species have no known fossil record, biogeographers often rely on reconstructing ancestral species ranges to understand how ranges change over time.
Only recently, parametric models have been used in a phylogenetic context to estimate ancestral species ranges, represented presence-absence states for sets of discrete areas.
Methodologically, we are interested in techniques to allow the models to be applied to high-resolution, global-scale datsets.
For models, we are interested how biogeographic inference may improve phylogenetic estimates.
|
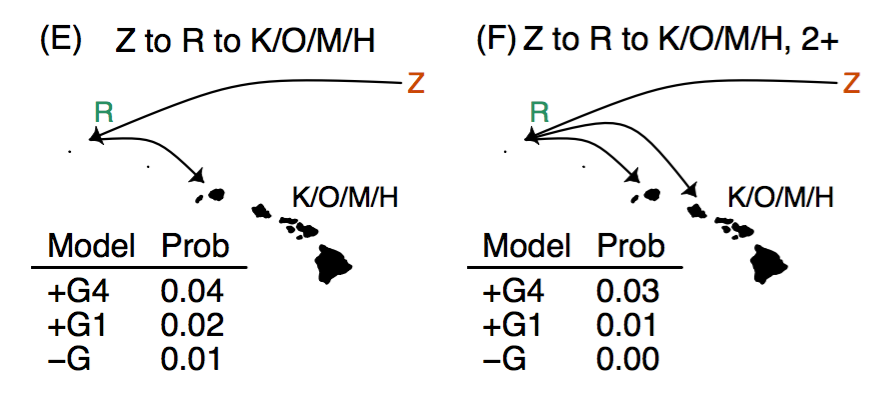
Landis, M. J., Freyman, W. A., & Baldwin, B. G. (2018). Retracing the silversword radiation despite phylogenetic, biogeographic, and paleogeographic uncertainty. Evolution (Accepted).
[paper] [scripts]
|
|
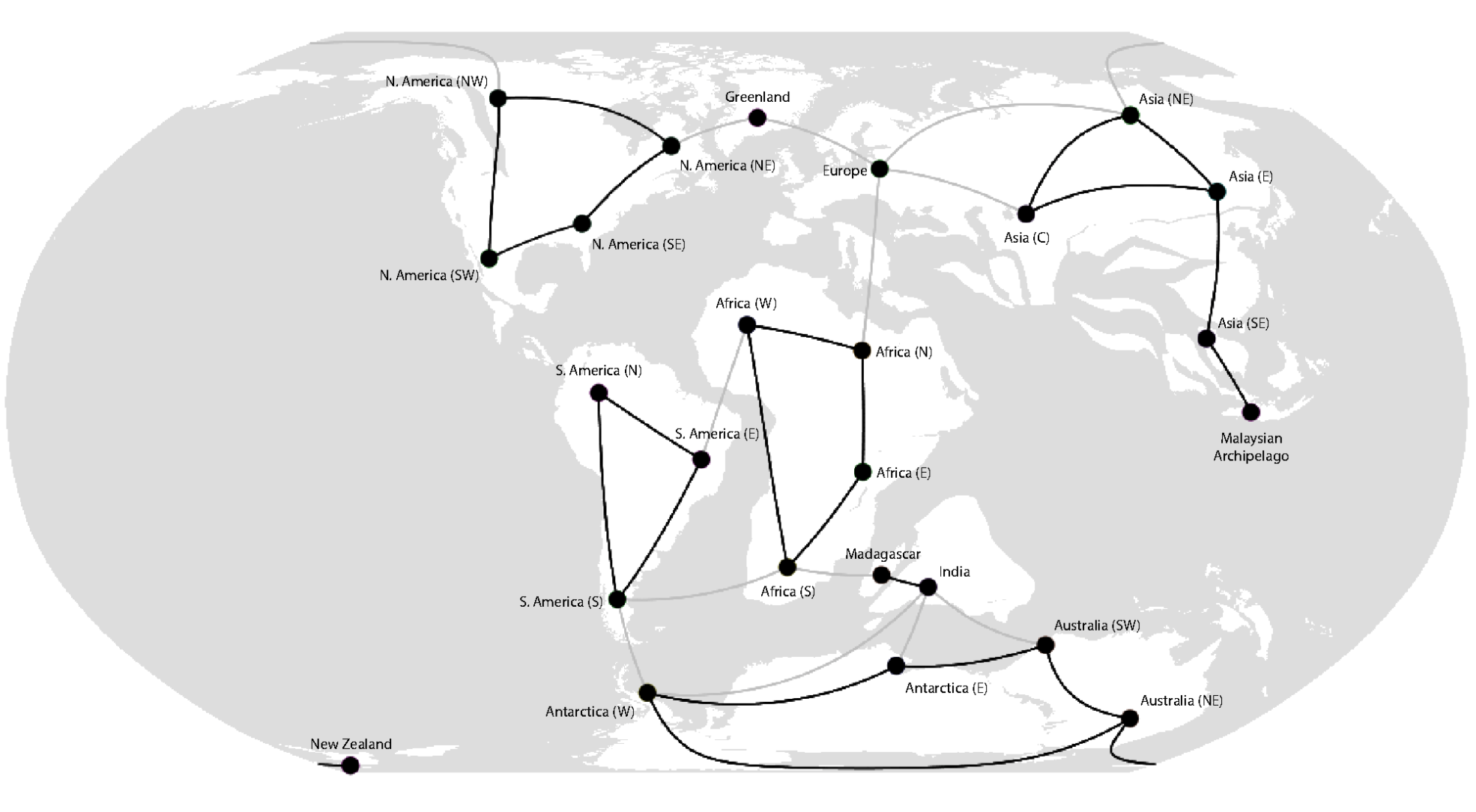
Landis, M. J. (2017). Biogeographic dating of speciation times using paleogeographically informed processes. Systematic Biology, 66(2):128-144.
[paper] [scripts]
|
|
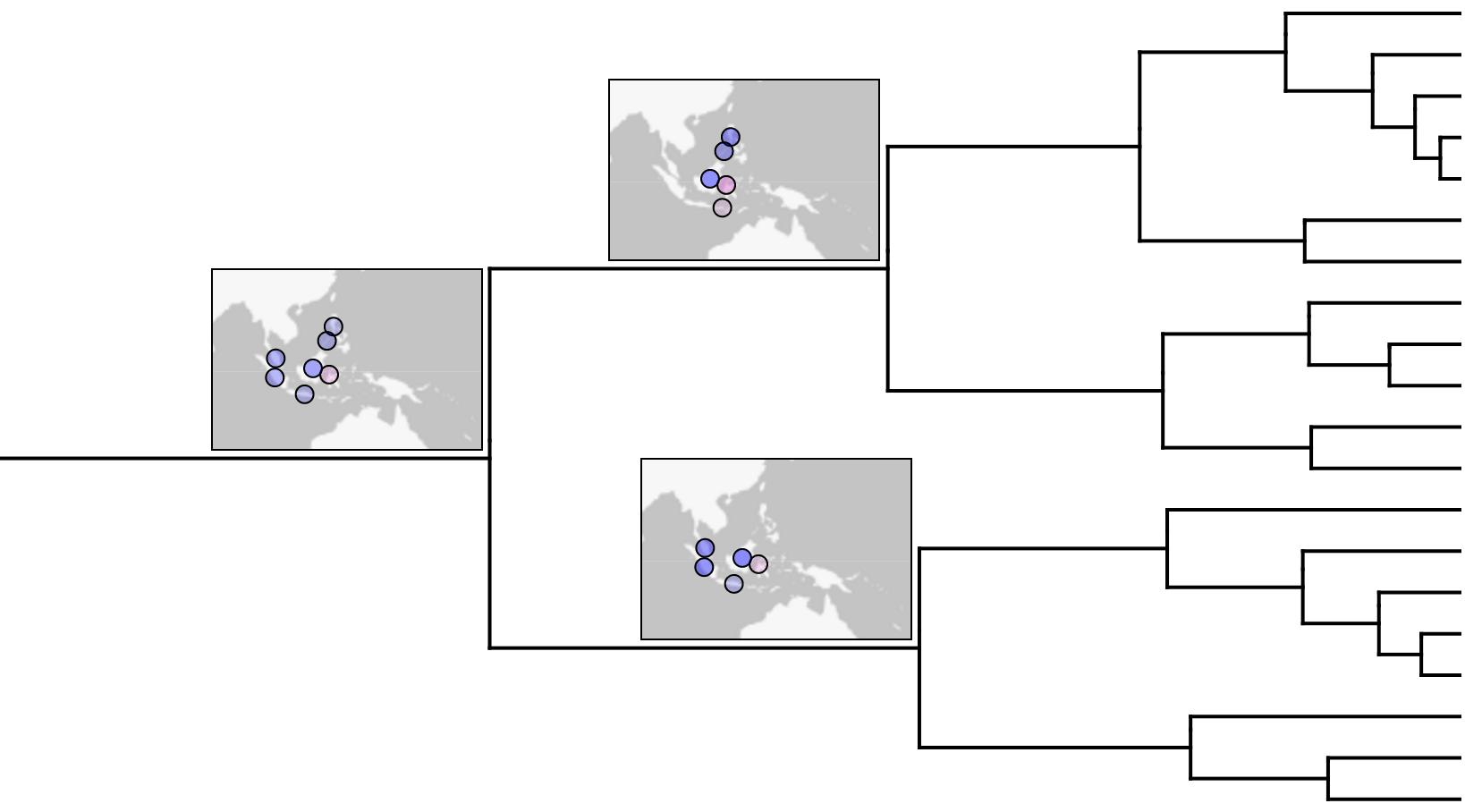
Landis, M. J., Matzke, N. J., Moore, B. R., & Huelsenbeck, J. P. (2013). Bayesian Analysis of Biogeography when the Number of Areas is Large. Systematic Biology, 62(6):789-804.
[paper] [software]
|
|
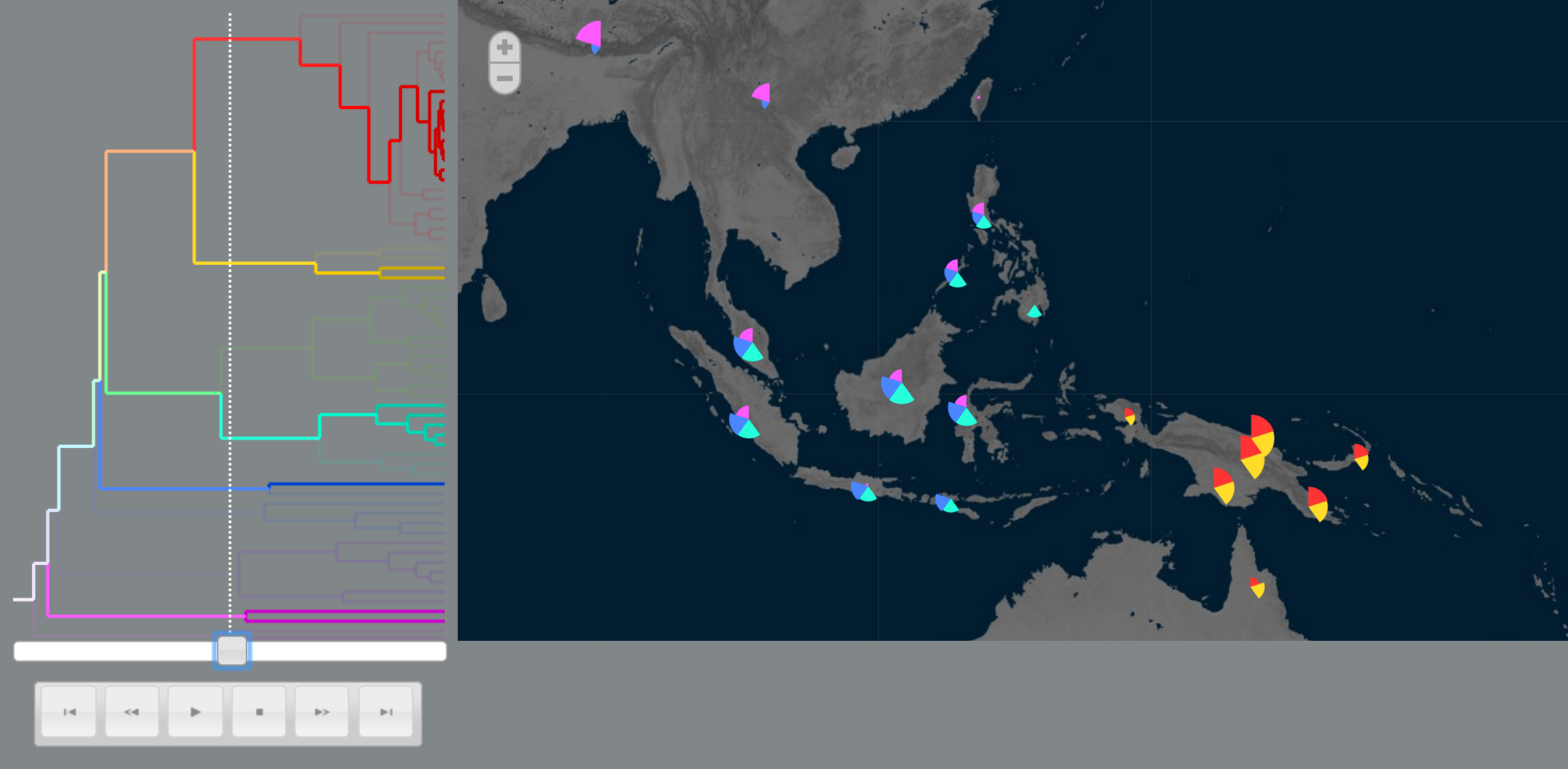
Landis, M. J. and Bedford, T. (2014). Phylowood: interactive web-based animations of biogeographic and phylogeographic histories. Bioinformatics, 30(1), 123-124.
[paper] [software]
|
Phenotypic evolution
Darwin’s original conception of evolution proposed that species evolve gradually over time.
In phylogenetics, this is often modeled as a Brownian motion, whereby small changes occur over many small intervals of time.
Many evolutionary mechanisms, such as rapid adaptation, may produce “pulses” in variation, with punctuated change of large effect.
Rapid change should produce power-law (or heavy-tailed) distributions of traits, but Brownian motion does not generate this feature.
It is difficult to detect these bursts if they are not modeled appropriately.
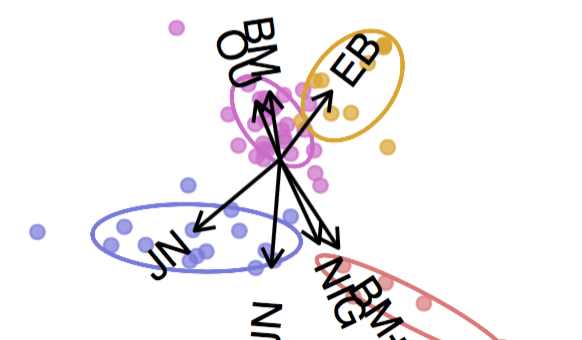
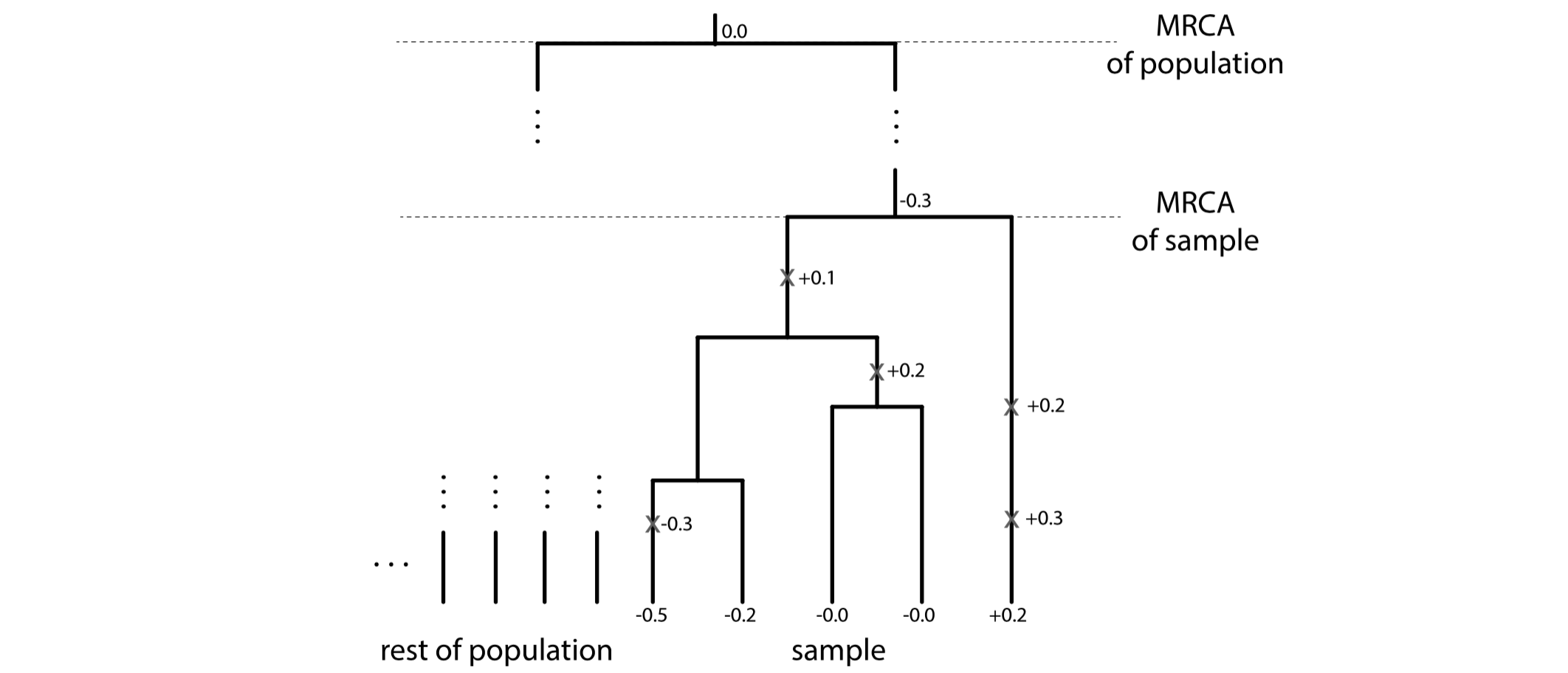
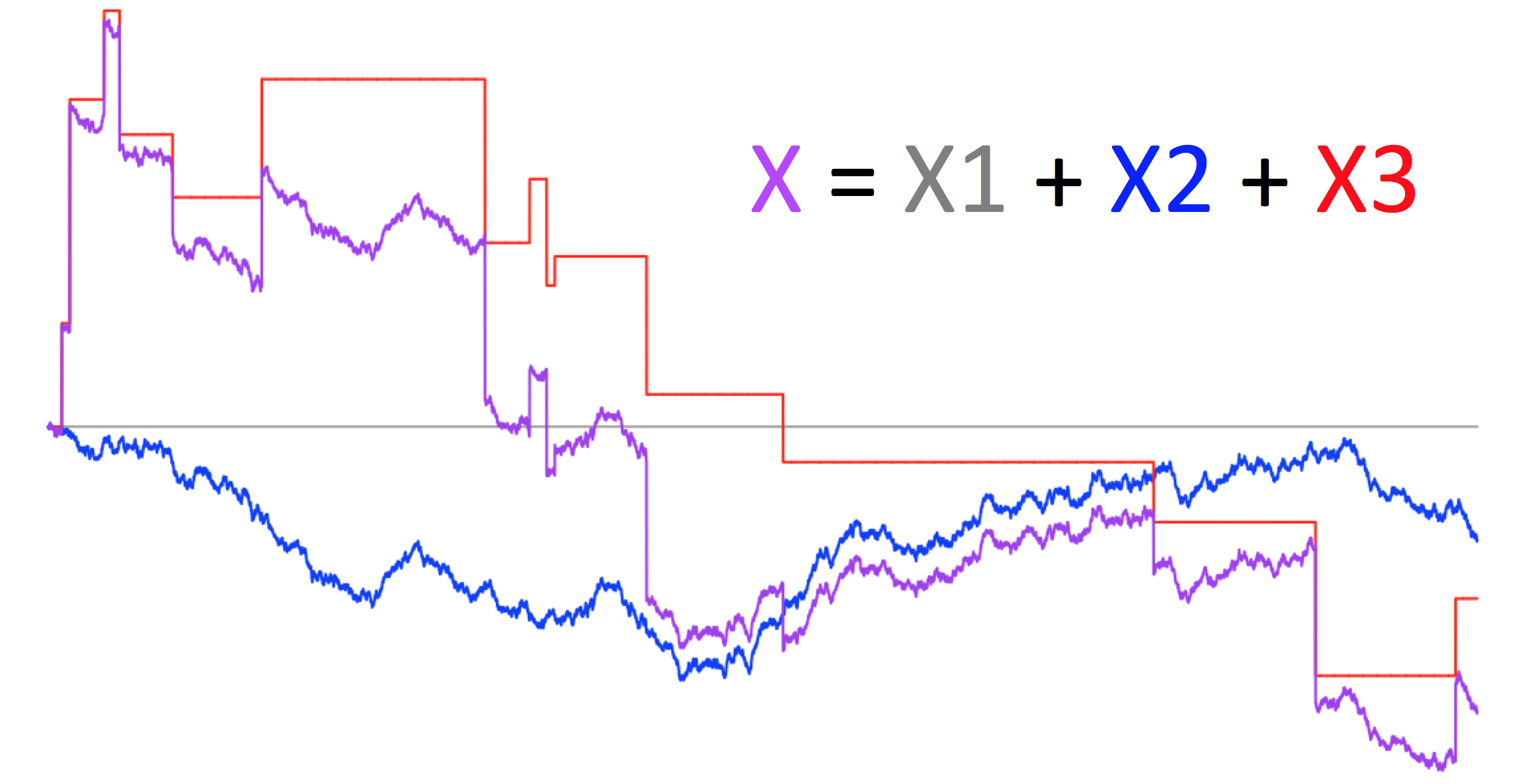
To search for signals of punctuated evolution, we apply a flexible class of stochastic processes that produce gradual and/or punctuational patterns of change, called Lévy processes.
|
Landis, M. J. and Schraiber, J. G. (2017). Pulsed evolution shaped modern vertebrate body sizes. Proceedings of the National Academy of Sciences, 114(50) 13224-13229.
[paper] [software]
|
|
Schraiber, J. G. and Landis, M. J.. (2015). Sensitivity of quantitative traits to mutational effects and number of loci. Theoretical population biology, 102: 85-93.
[paper] [scripts]
|
|
Landis, M. J. (*), Schraiber, J. G. (*), & Liang, M. (2013). Phylogenetic Analysis Using Lévy Processes: Finding Jumps in the Evolution of Continuous Traits. Systematic Biology, 62(2):193-204.
[paper] [software]
|
(*) - Lead co-authors.
Phylogenetic inference with RevBayes
Learning the degree of relatedness between species has far-reaching implications for biological research, from basic research questions in evolution and ecology, to applied questions in medical research, epidemiology, forensics, and biodiversity management.
Estimating a phylogeny, of course, depends on one’s model assumptions, which vary depending on the nature of the study.
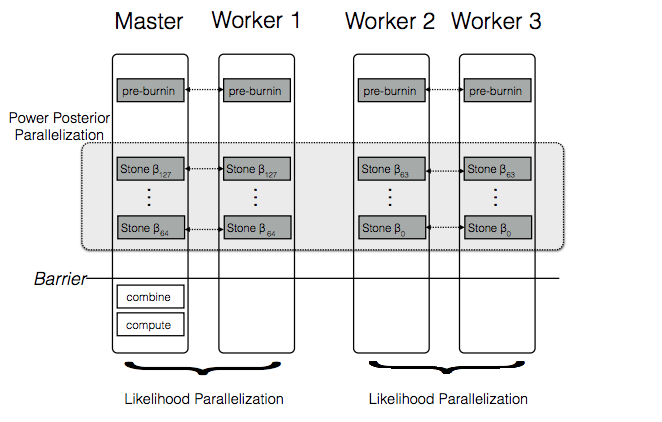
RevBayes was designed to allow researchers to tailor models to their needs.
RevBayes achieves this by providing a flexible scripting language to describe probabilistic graphical models.
|
Höhna, S., Landis, M. J., and B. R., Huelsenbeck. (2017). Parallel power posterior analyses for fast computation of marginal likelihoods in phylogenetics. Bioinformatics, Accepted.
[paper (pre-print)]
|

|
Höhna, S., Landis, M. J., and Heath, T. A. (2017). Phylogenetic Inference Using RevBayes. Current Protocols in Bioinformatics, 57:6.16-6.16.34.
[paper]
|
|
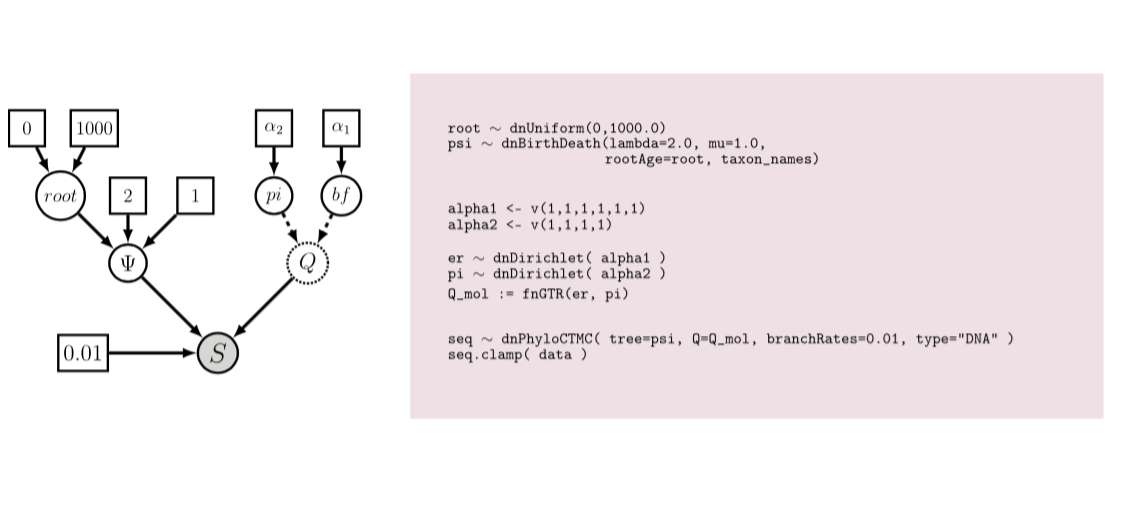
Höhna, S., Landis, M. J., Heath, T. A., Boussau, B., Lartillot, N., Moore, B. R., Huelsenbeck, J. P. & Ronquist, F. (2016). RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Systematic Biology, 65(4):726-736.
[paper] [software]
|
|
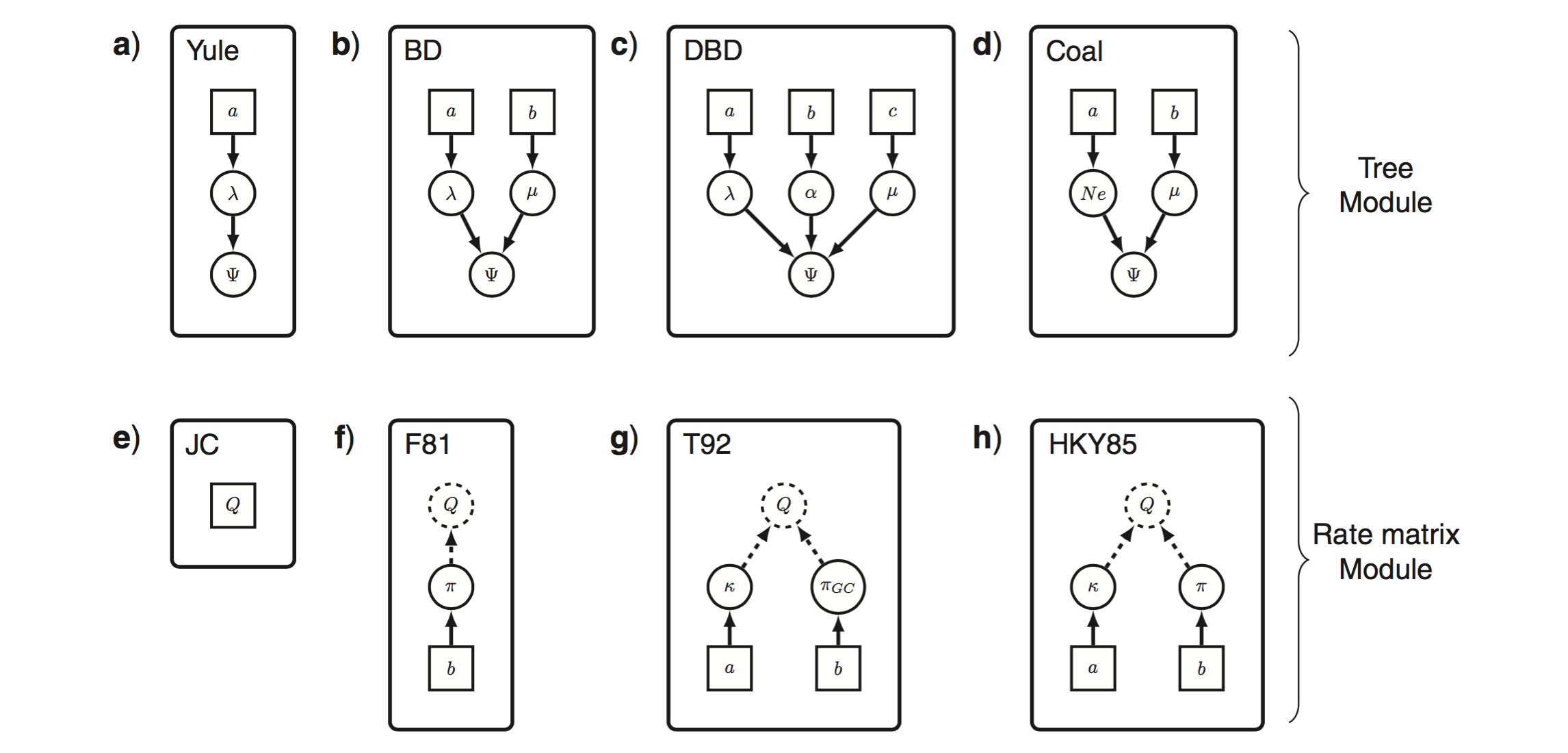
Höhna, S., Heath, T. A., Boussau, B., Landis, M. J., Ronquist, F., & Huelsenbeck, J. P. (2014). Probabilistic graphical model representation in phylogenetics. Systematic Biology, 63(5):753-771.
[paper]
|